Central Role of Amyloid Oligomers in Alzheimer’s
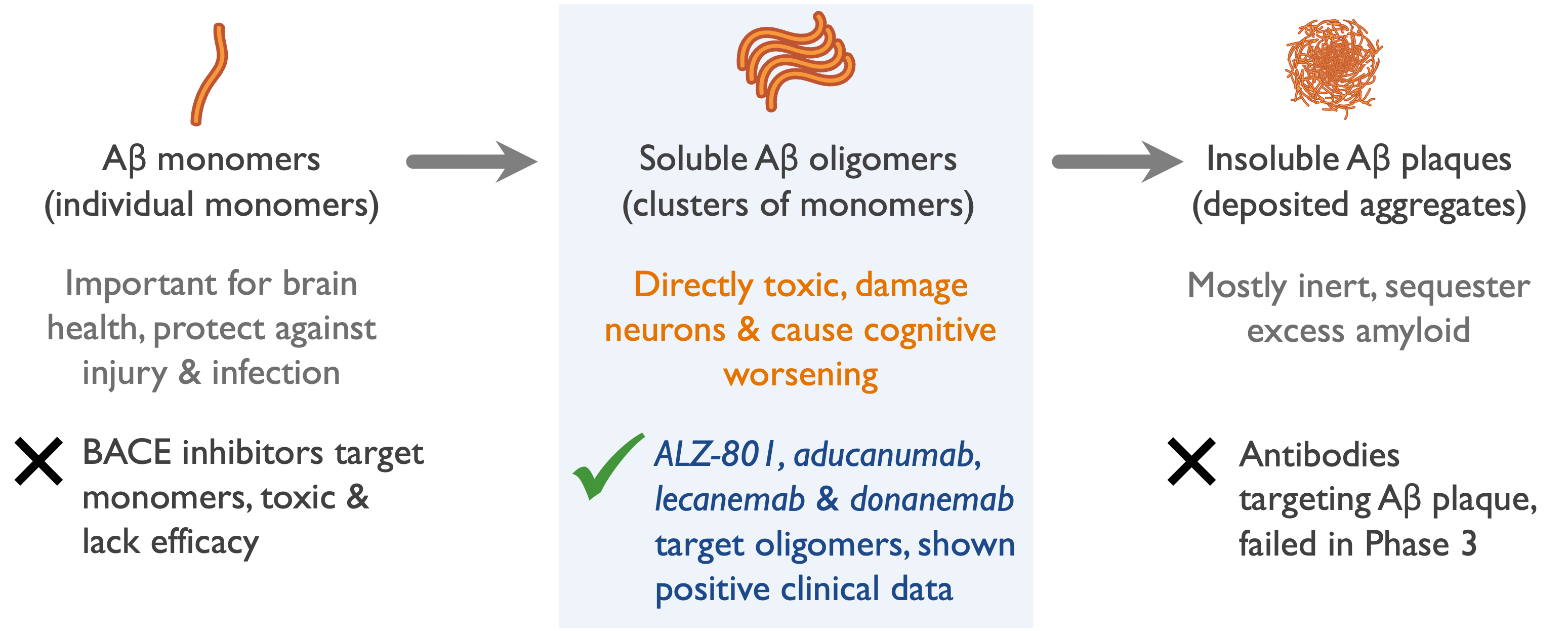
A large body of scientific evidence suggests that neurotoxic soluble amyloid oligomers trigger neuronal damage and cell death leading to Alzheimer’s disease (AD).
ALZ-801/valiltramiprosate is designed to inhibit amyloid oligomer formation
Our lead product candidate, ALZ-801, is designed to inhibit amyloid oligomer formation, a key driver of Alzheimer’s disease. We believe ALZ-801 has the potential to be differentiated from other emerging therapies targeting Alzheimer’s disease pathology due to its novel mechanism of action, oral mode of administration and potential efficacy in a genetically-targeted population. If our development program is successful and ALZ-801 is approved, it has the potential to be among the first drugs to intervene in an underlying mechanism of Alzheimer’s disease.
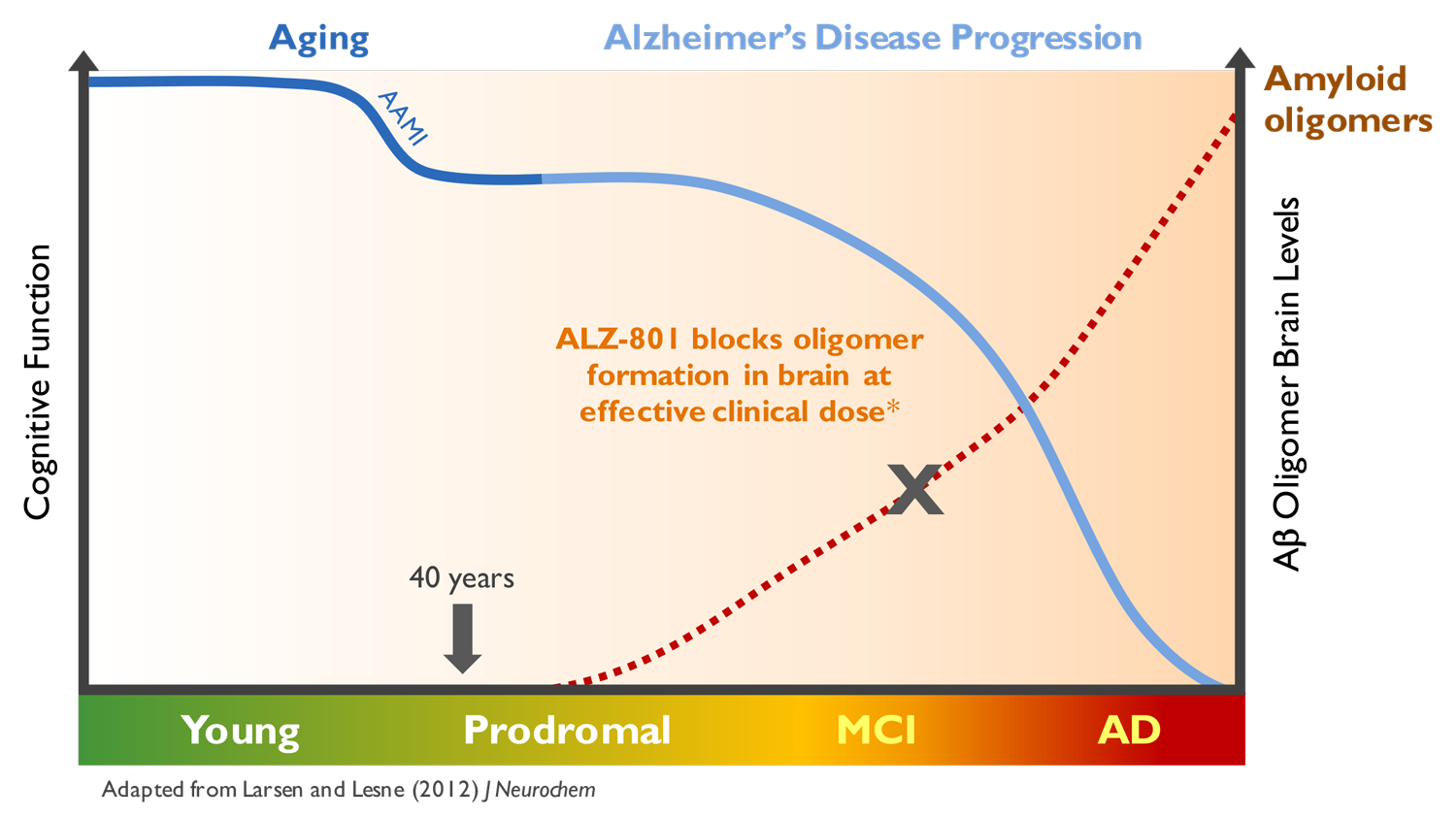
Neurotoxic soluble amyloid oligomers present early in AD
- Soluble amyloid oligomers damage neurons & synapses
- Oligomer levels in brain increase & drive clinical progression
- APOE4/4 homozygotes form more oligomers leading to an earlier disease onset
- Targeting oligomers in AD patients supported by clinical benefits in antibody programs
Alzheon publications: Kocis (2017) CNS Drugs; Hey (2018) Clin Pharmacokinet; Hey (2018) CNS Drugs
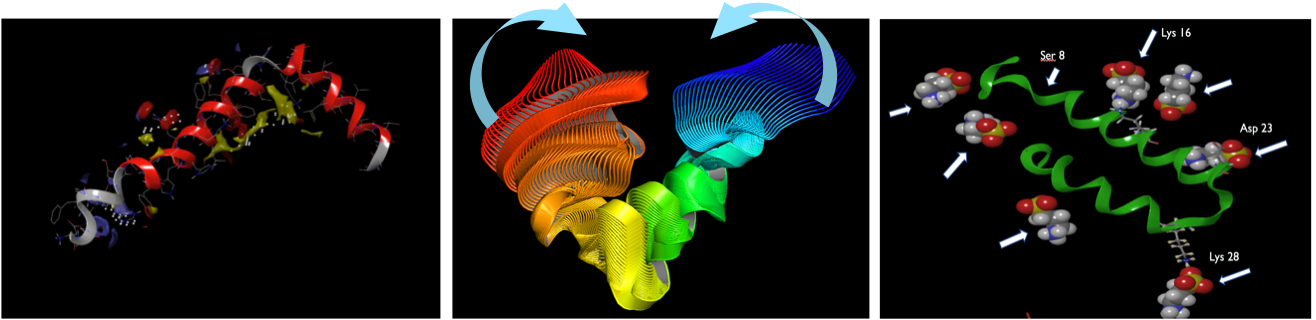
Enveloping mechanism of action of ALZ-801
Multiple molecules bind to amyloid & prevent toxic oligomer formation
- Several molecules surround & interact with beta amyloid monomers
- Stabilize shape of amyloid monomers & block formation of neurotoxic oligomers
- Interact with key amino acids on beta amyloid monomer
Alzheon publications: Hey (2018) CNS Drugs; Kocis (2017) CNS Drugs
ALZ-801 protects native state of beta amyloid protein
Multiple molecules of ALZ-801/tramiprosate form an enveloping cloud around Aβ42 amyloid monomer that maintains its native shape & prevents aggregation into toxic oligomers
Amyloid conformation enforced by ALZ-801
Influenced by surrounding cloud of excess ALZ-801 molecules, Aβ42 amyloid monomer adopts shape that blocks formation of oligomers
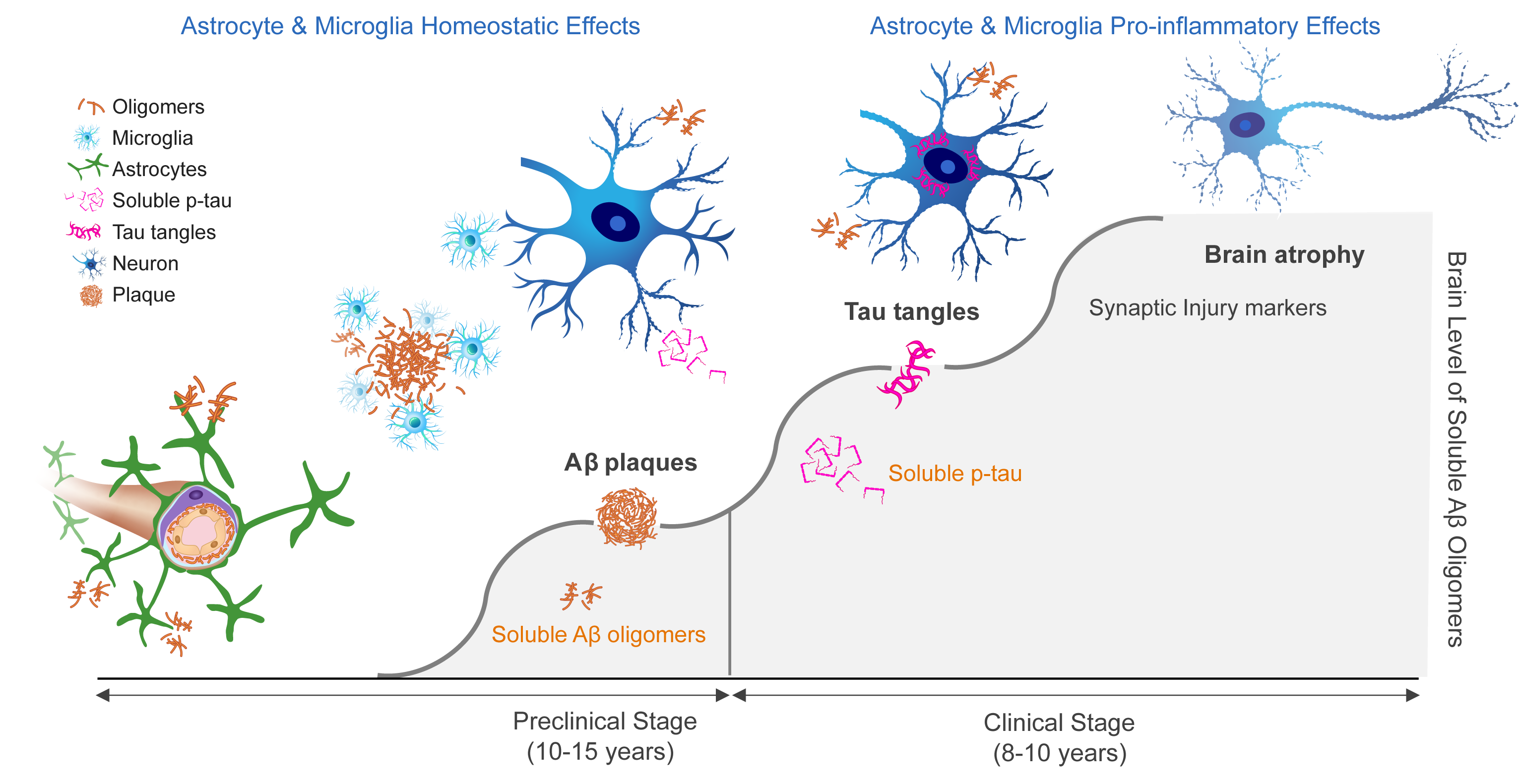
Aβ oligomer toxicity across stages of Alzheimer’s disease