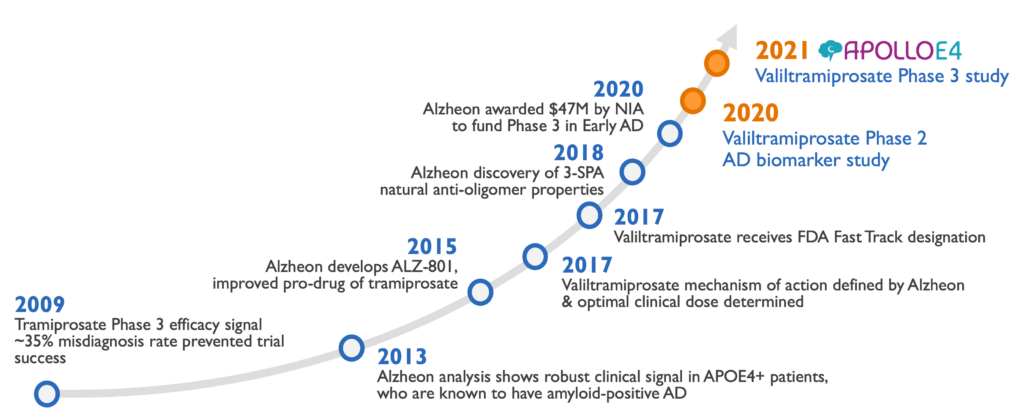
Phase 3 study evaluated efficacy of valiltramiprosate in APOE4/4 Early AD subjects
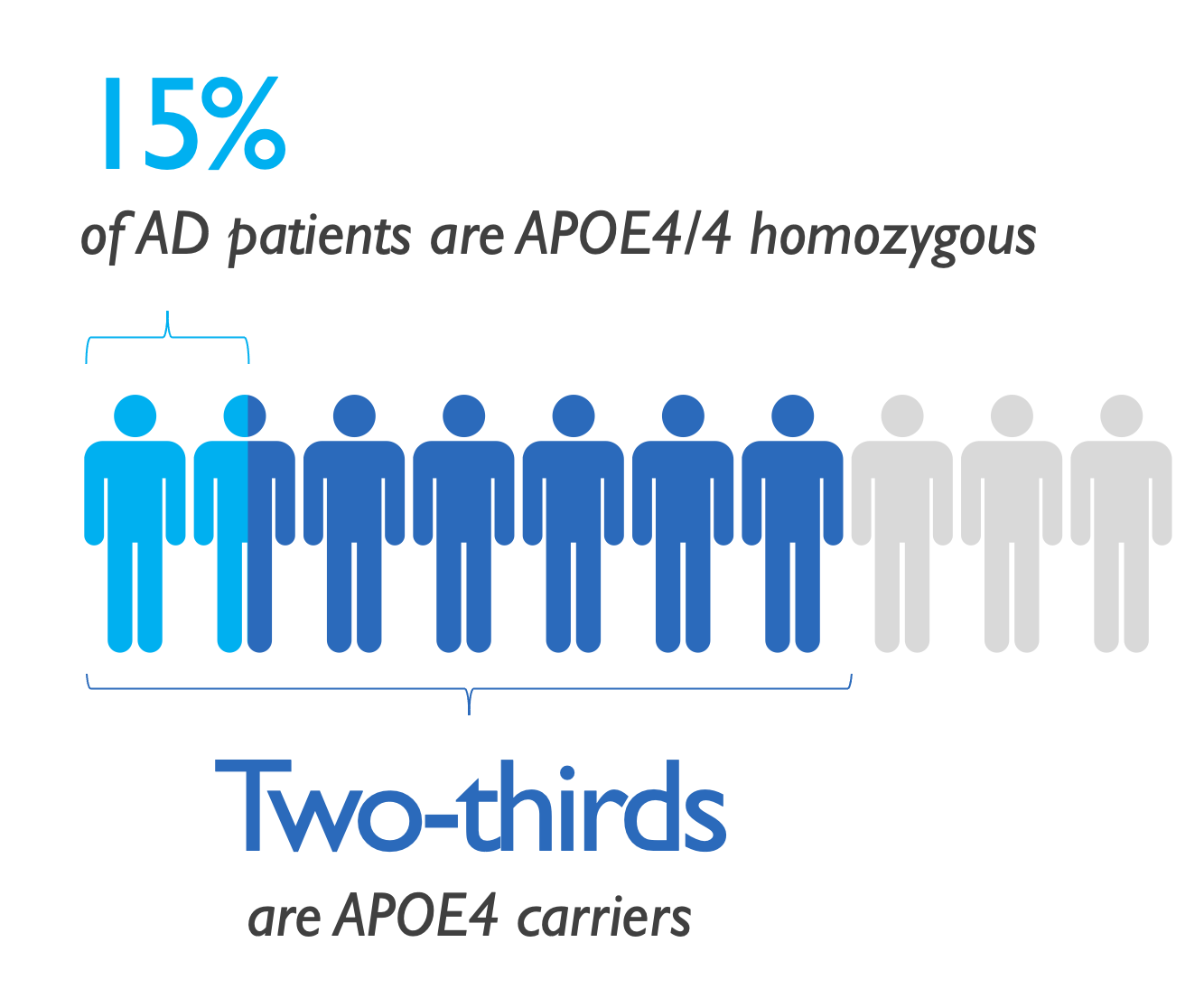
Valiltramiprosate Phase 3 trial in APOE4/4 patients with Early AD
APOLLOE4 pivotal trial enrolled 325 subjects
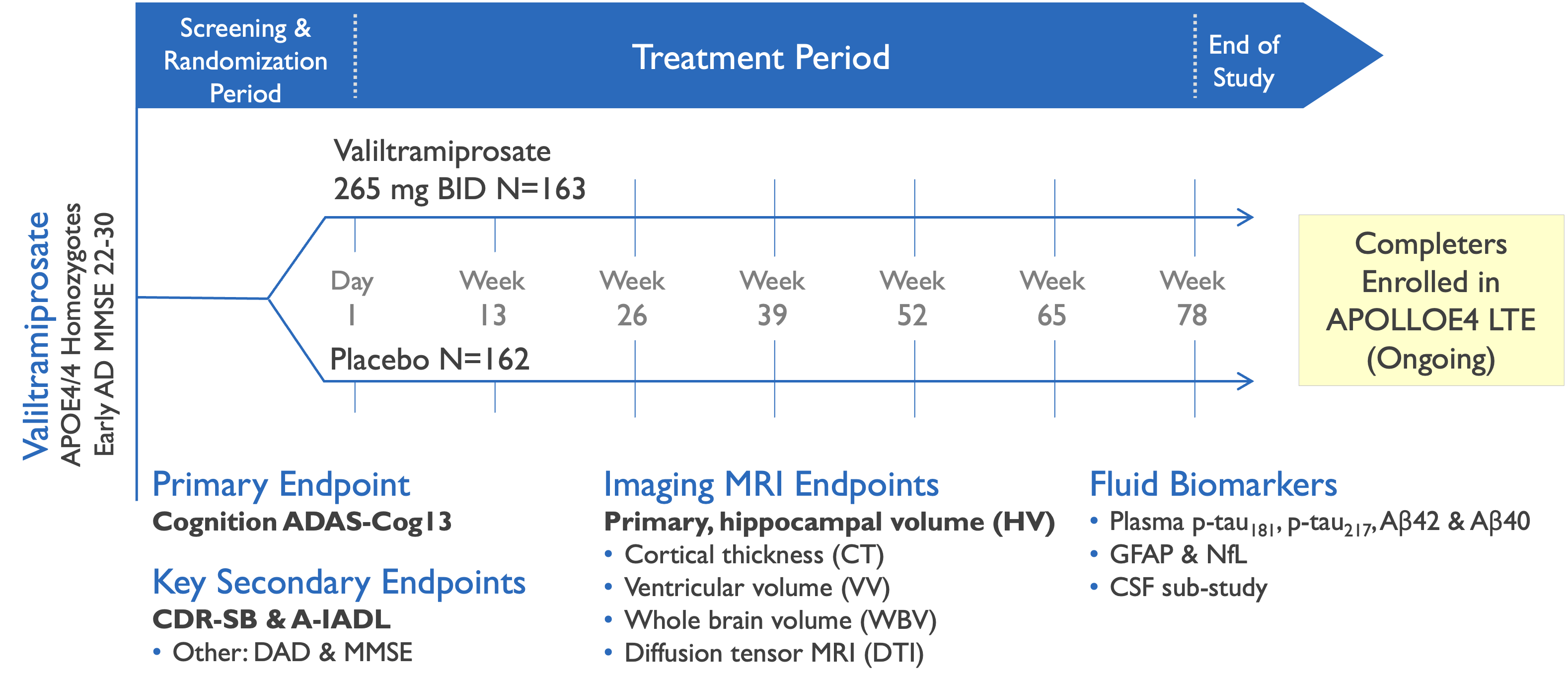
Valiltramiprosate development history